Understanding Hunter syndrome
What is Hunter syndrome?1,2
Hunter syndrome, or mucopolysaccharidosis type II (MPS II), is a rare progressively debilitating genetic disorder in which large sugar molecules called glycosaminoglycans (also known as GAGs, or mucopolysaccharides) build up in brain and body tissues. Hunter syndrome is caused by a deficiency of the lysosomal enzyme iduronate-2-sulfatase, which is responsible for the breakdown of glycosaminoglycans.
What are the signs and symptoms of Hunter syndrome?1,2
Though the disease is present at birth, symptoms become more apparent as the accumulation of GAGs builds within the body. Typically, children continue to develop physically and cognitively until between the ages 2-5, and then they may begin regressing.
Traditionally, Hunter syndrome is described as having two forms:
- Attenuated form—with fewer cognitive impairments and slower progression, affecting approximately one-third of individuals diagnosed with Hunter syndrome
- Neuronopathic form—impacting cognitive aspects of the child, affecting approximately two-thirds of individuals diagnosed with Hunter syndrome
However, it is understood that there is a wide spectrum of disease severity in how MPS II impacts each child’s progression.
Features and symptoms of Hunter syndrome may include:
-
Carpal tunnel syndrome
-
Developmental delay
-
Disruptive behaviors
-
Hearing loss
-
Hydrocephalus (water on the brain)
-
Impaired cognition
-
Impaired sleep
-
Macrocephaly (large head)
-
Recurrent otitis media (ear infection)
-
Reduced pulmonary function
-
Reduced range of joint motion
-
Spinal stenosis
-
Valvular heart disease
-
Vision loss
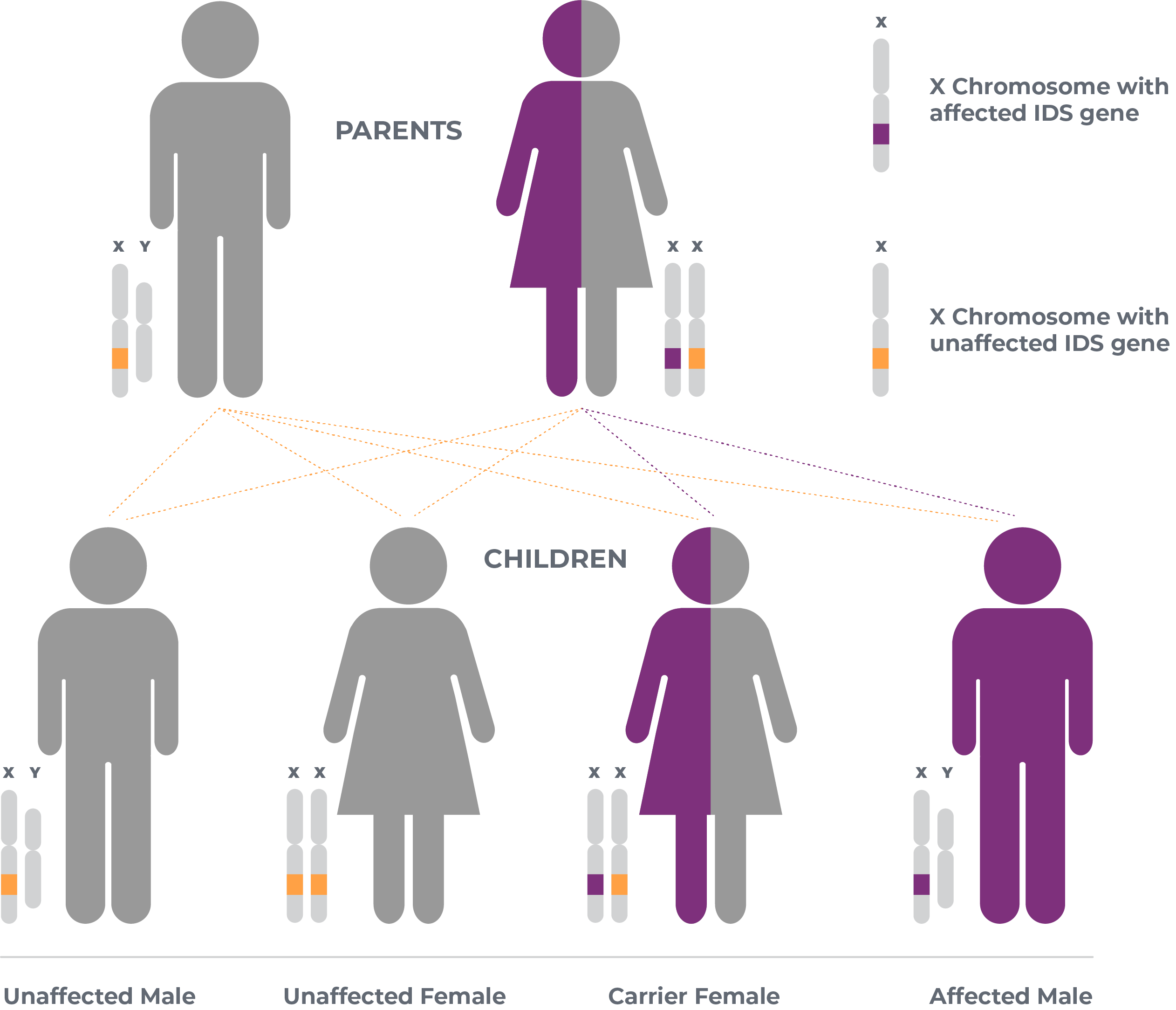
What is the link between Hunter syndrome and genetics?2
Hunter syndrome is an X-linked recessive genetic disease. This means that women who carry the chromosome with the disease causing gene can pass it on to their children. Families should speak with their healthcare provider for more information.
What is the iduronate-2-sulfatase (IDS) gene?4
The IDS gene provides instructions for producing an enzyme called iduronate-2-sulfatase, which is essential for the breakdown of large sugar molecules called glycosaminoglycans (GAGs). Specifically, iduronate-2-sulfatase removes a chemical group known as a sulfate from a molecule called sulfated alpha-L-iduronic acid, which is present in two GAGs called heparan sulfate and dermatan sulfate. The iduronate-2-sulfatase enzyme is located in lysosomes, compartments within cells that digest and recycle different types of molecules. When this enzyme is deficient, GAGs build up in the lysosomes and cause them to malfunction, leading to the symptoms of Hunter syndrome. The current standard treatment for Hunter syndrome, IV enzyme replacement therapy, treats the peripheral symptoms of the disease but does not appear to impact the neurocognitive components of the disease.
Learn More About Clinical Trials
Whether you’re interested in active participation or just want to know about research in progress, see our latest clinical trial information here
References: 1. https://www.mayoclinic.org/diseases-conditions/hunter-syndrome/symptoms-causes/syc-20350706 Accessed 07/06/2020. 2. Scarpa M. Mucopolysaccharidosis Type II. 2007 Nov 6 [Updated 2018 Oct 4]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. 3. Ullman et al., Sci. Transl. Med. 12, eaay1163 (2020) 27 May 2020 4. https://ghr.nlm.nih.gov/gene/IDS Accessed 07/06/2020.